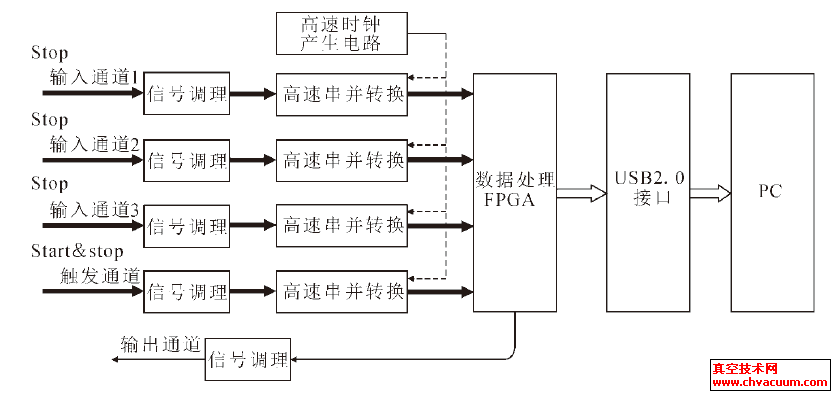
����Û���x���Ƃ�ʯīϩ�������
������-48μm���[Ƭʯī��ԭ�ϣ��Ȳ���Hummers���Ƃ�����ʯī���ٲ��øߜ�?z��)���Û���x���Ƃ�ʯīϩ������X�侀����(XRD)�������~�t����V(FT-IR)��ԭ�����@�R(AFM)��N2 ����-Ó��(BET)���о�������ʯī��ʯīϩ�ľ��w�Y(ji��)��(g��u)��������܈F��������ò���ȱ���e�����ֲ��ȡ�XRD �о��Y(ji��)������������ʯī���g����0.94 nm��ԭ�е�ʯī����ʧ;����Û����ʯīϩ(2θ=25.6°��d(002)=0.348nm)��o���ΑB(t��i)��FT-IR ����������ʯī�����^���нY(ji��)��(g��u)���g�γɴ����������܈F����(j��ng)�ߜ�߀ԭ��H���沿�ֺ������܈F��ʯīϩ�����^�ߵıȱ���e(336.7m2/g)��������0.4~0.7 nm ֮�g����1~2 ��ʯīϩ��
����2004 �꣬Ӣ������˹�D��W(xu��)K S Novoselov ��A K Geim ���ˣ��ڌ����ͨ�^�z������(f��)���xʯīƬ�l(f��)�F(xi��n)��ֻ��1 ��ԭ�Ӻ�Ȇξ�ʯīĤ——ʯīϩ��ʯīϩ���Ͼ�����Փ�߱ȱ���e (2600 m2/g) �Լ����ص������(15000cm2/(V·s))����(d��o)������(3000 W/(m·K))������ģ��(1.01 TPa)���O�ޏ���(116 GPa) ��W(xu��)���|(zh��)�������˿ƌW(xu��)�ҵďV���P(gu��n)ע��
����Ŀǰ��ʯīϩ���Ƃ䷽����Ҫ�֞黯�W(xu��)���������������W(xu��)����������Û���x�������W(xu��)������e��������ʯī߀ԭ����늻��W(xu��)����ʯī��ӷ��ȡ������������Cе���x������ը�����ӟ�SiC ����ȡ��������ʯīϩ��ͨ�^��Ûʯī��������ĥ�Ƃ䣬��Ƭ�Ӻ��һ���30~100 nm���y�Եõ��Ό�ʯīϩ����������Ȳ���Hummers ���Ƃ�����ʯī���� 1050 ��ߜ�?z��)���Û����ͨ�^��ˮ��Һ�г��Ƃ���1~2 ��ʯīϩ��
1������
1.1��ԭ�ϼ�ԇ��
������Ȼ���[Ƭʯī����̼99.99%��������-48μm����X �侀�����������(002)�����g����0.336 nm�����i��⛡�98% �����ᡢ�����c��30%�p��ˮ��5% �}�ᣬ�����������
1.2������
��������ʯī�Ƃ䣺����Hummers ���Ƃ�����ʯī�������ڸ�������м���55 mL 98%�������1 g �����c����ԡ�l������s����(d��ng)�wϵ�ضȵ���5 ��r�������м���2 g �[Ƭʯī����Ͼ����������5 g ���i��⛣����Ʒ���(y��ng)Һ�ضȲ����^ 20 �棬����(y��ng)2 h��Ȼ�������� 35 �����ҵĺ��ˮԡ�У����裬�����Һ�ض����� 35 �棬����(y��ng)30 min������92 mL ȥ�x��ˮ�����Ʒ���(y��ng)Һ�ض��� 98 �����ң��^�m(x��)����15 min��Ȼ�����280 mL ȥ�x��ˮ������(y��ng)�Kֹ��ͬ�r����20 mL 30% �p��ˮ���@�r��Һ���غ�ɫ׃?y��u)��r�����Sɫ���ß��^�V������2 L 5% ϡ�}�ጦ�a(ch��n)���M��ϴ�죬��ȥ�x��ˮ���ϴ��ֱ��pH ֵ�����ԡ�Ȼ����60 ������и����ĥ�����@�õ�����ʯī���ڸ������б��档
����ʯīϩ�Ƃ䣺ȡ0.1 g ����ʯī��ĩ����100 mL�մ�����У��ü��F�z���w�ӽ��ù̶��������õ�������1050 ���R���t�У�30 s ��ȡ�������ÿɄ��xʯī���Ʉ��xʯī��1 mg/mL ���뵽ˮ��Һ�У���̎��20 min���õ�ʯīϩ�Ҹ�Һ���������õ�ʯīϩ��
1.3�����ܱ���
����XRD ����ʹ�õ��|DX-2700 ��X�侀����x����Cu-Kα (40 kV��40 mA) ���侀Դ�����跶��3��~80�������M����0.02°�����M����10 (��)/min��FT-IR �yԇ�������������˾Nexus-670�����~׃�Q�t����V�x����ɣ��ƘӲ���KBr ��Ƭ���yԇ����(sh��)������400~4000 cm-1��AFM ԭ�����@�R���ڱ�������ʯī��ʯīϩ�ļ{��Ƭ��ȼ��Ӕ�(sh��)�����������S�ƹ�˾3D �{���@�R�p��ģʽ�M�Мyԇ��
�����yԇ��Ʒ�nj���ˮ��Һ�г���Ę�Ʒ�Ҹ�Һ�A(y��)�ȵ�����ĸ���棬��Ȼ�����Ƴɡ���������������˾Autosorb-1 �ͱȱ���e�����ֲ������x��ʯīϩ�M��BET��������(bi��o)��(zh��n)��≺��ʹ�õ�������- Ó�����o�B(t��i)�������y����
2���Y(ji��)���cӑՓ
2.1��X �侀�������
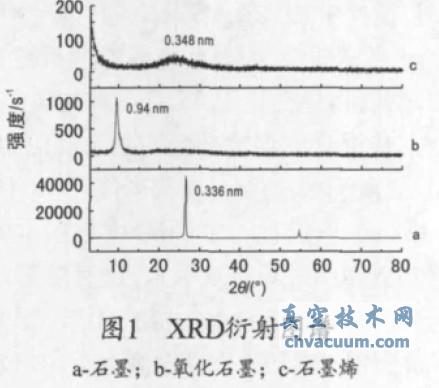
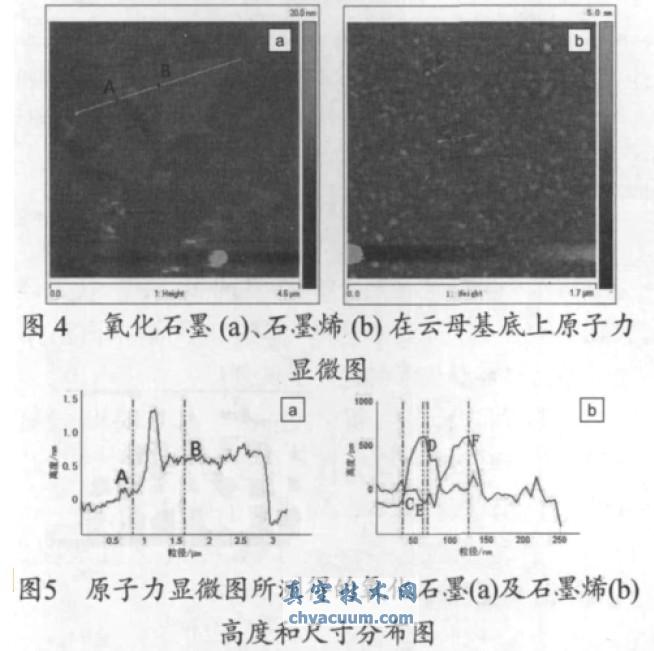
����ʯīԭ�ϡ�����ʯī�Լ�ʯīϩ��XRD ������D��Ҋ�D1���ĈD1a �ɿ�����ʯī(002) �������2θ ��26.5��̎�dz����J������(y��ng)���g����0.336 nm���f����ʯī���̶ȸߣ���Ƭ�ӵĿ��g���зdz�Ҏ(gu��)��;2θ ��54.6��̎��ʯī(004) ��������塣�ĈD1b �ɿ�����ʯī��������ʯī(002) ��������������ʧ������ʯī�Ӡ�Y(ji��)��(g��u)�����������(001)���F(xi��n)��9.4°̎�����g����0.94 nm�����g��õ������ߣ����g�����A���pС������ͨ�^����Û�õ�ʯīϩ������g���h����ʯī���g��(0.336 nm)���@�w��������ʯī���g��������������܈F(�u�����Ȼ����h(hu��n)������)���ĈD1c �ɿ�������(d��ng)����ʯī��(j��ng)�^�ߜ���Û߀ԭ������ʯīλ��9.4��̎���������ʧ����ʯī���ķ�l(f��)�������������^����2θ ��25.6°��d(002) ��0.348 nm�f��ʯī���g�ѽ�(j��ng)�a(ch��n)���˟ᄃ�x��߀ԭ��ʯīƬ�ӳߴ�pС�����w�Y(ji��)��(g��u)�������½����o������ӣ��õ��o���͑B(t��i)ʯīϩ��
2.2���t����V����
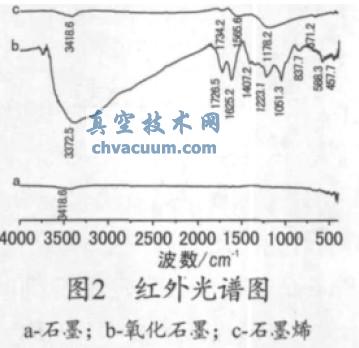
����ʯī������ʯī�Լ�ʯīϩ�t��D�V��Ҋ�D2���ĈD2 �ɿ�����ʯī�����o�t�����շ壬����ʯī���g���^�ຬ�����܈F����(j��ng)�ߜ�߀ԭ���@Щ���܈F������ʧ��p�٣�����(y��ng)�ļt��������շ�Ҳ����ʧ��p�����D2b ������ʯī�ڸ��l�^(q��)3372 cm-1 �������^�������շ壬�w����O-H ��s��ӣ���2000~3700 cm-1 �������F(xi��n)�^�����V�壬����������ʯī�����Ĵ���ˮ���ӵ���s��ӣ������l�^(q��)1726 cm-1 ̎���F(xi��n)�����շ壬�w��������ʯī߅���Ȼ����ʻ���C=O ��s���[7]��1652 cm-1 ̎���շ�w��������ʯī���gˮ����O-H ����Ӻ�δ����ʯīsp2 ̼�Ǽ�C=C ����ӣ�λ��1407 cm-1 ̎�����շ�w����-H ׃�����[9]��1223 cm-1 ������C-OH ����������շ�Ĝp����1051 cm-1 ̎�����շ�w����C-O-C ��ӡ��@Щ�������F�Ĵ����f����ʯī�������������u���Ĵ���ʹ������ʯī�����cˮ�����γɚ��I�����F(xi��n)���ܺõ��Hˮ�ԡ��ĈD2c �ɿ�����λ��3372 cm-1̎O-H �����շ����@�p����λ��1565 cm-1 ̎�w����ʯīϩƬ�ӹǼ�-C=C- ��ӵ����շ���u������ͬ�rλ��1178 cm-1 ̎�w���ڭh(hu��n)�����F�����շ����@�^����1051 cm-1 ̎���շ匦��(y��ng)�� C-O-C �I�ķ����Q�����u�p�����@Щ���C��������ʯī��߀ԭ��
2.3������ʯī���@�Y(ji��)��(g��u)��ʯīϩԭ��
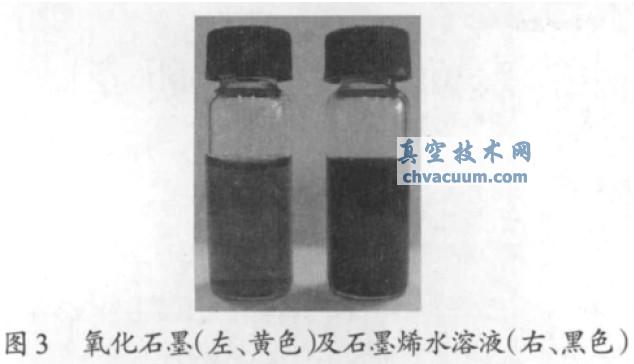
��������ԭ�����@�R��������ʯī��ʯīϩ��ò��ˮ��Һ�г��õ�������ʯī��ʯīϩ��Һ�ڔ�(sh��)�܃�(n��i)���ַ�(w��n)�������l(f��)������(�D3)������ʯī��ʯīϩ����ĸ������ԭ�����@�D�Լ�ԭ�����@�D���y�õ�����ʯī��ʯīϩ�߶Ⱥͳߴ�ֲ��D���քeҊ�D4���D5���ĈD4a���D5a �ɿ���������ʯī߅���l(f��)�������دB������ʯī�����ֲ��^������ƽ����ȴ�s1 nm���@�cXRD��������ʯī���g����0.94 nm ����һ�£������ѽ�(j��ng)�F(xi��n)��ȫ���x��ʯī������������ʯīƬ�Ӄ������й��r��ԭ�Ӻ�sp3 �s��̼ԭ�Ӷ�����ʹ���h����ʯīϩ���0.348 nm���ĈD4b���D5b �ɿ�����ʯīϩƬ���Ⱦ���ֲ�����ĸƬ���棬����ʯī���ڸߜ�߀ԭ���Ì�(d��o)�½Y(ji��)��(g��u)��(n��i)�����W(xu��)�I���ѣ����ȜpС��ʯīϩ�����0.4~0.7 nm ֮�g����1~2 ��ʯīϩ���D��CD ��EF ������һ�����ӳߴ��ʯīϩ���ĈD5b �߶������пɿ�����ߴ��s�ڎ�ʮ�{�ס�����ʯī��߀ԭ��ʯīϩ���ȜpС�@һ�F(xi��n)���c�����x�ȵ�ʯīϩ�z��Ҹ�Һ�������ȷ���һ�¡�
2.4��ʯīϩ�ȱ���e
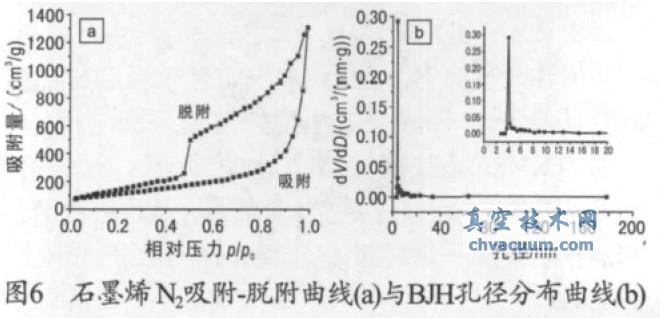
����ʯīϩ���ϵ�N2 ����- Ó�����������ֲ��D��Ҋ�D6���ĈD6a �ɿ�������Ʒ���м{�Y(ji��)��(g��u);�ĈD6b �ɿ�������Ʒ���к܌��Ŀ��ֲ���������3~180 nm������Ҫ������3~5 nm��ʯīϩ��BET�ȱ���e��336.7 m2/g�������ݞ�1.76 mL/g��ƽ������20.94 nm����Ʒ�ıȱ���e�ȆΌ�ʯīϩ�o��Ƭ��Փֵ2630 m2/g �ͣ��@��Ҫ���ܵ�ʯīϩ���ȵ������Լ�����ʯī�����̶Ȳ���������ȫ���x����Ʒ��(n��i)������Ƭ�����¶ѷe��
3���Y(ji��)Փ
��������Ȼ�[Ƭʯī��ԭ�ϣ�����Hummers ���Ƃ�����ʯī��ͨ�^�ߜ�?z��)���Û�õ��Ʉ��xʯī����̎���õ�ʯīϩ�{��Ƭ������ʯī����sp3 �s��̼ԭ�ӱ�߀ԭ��ʯī��sp2 �s����ʯīϩ���w�Y(ji��)��(g��u)�������½����o������ӡ�N2 ����- Ó��������ʯīϩ�����^�ߵıȱ���e����336.7 m2/g��AFM �yԇ����ˮ��Һ����õ���1~2 ��ʯīϩ��