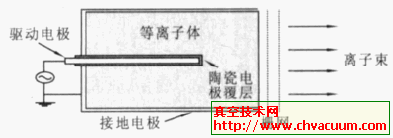
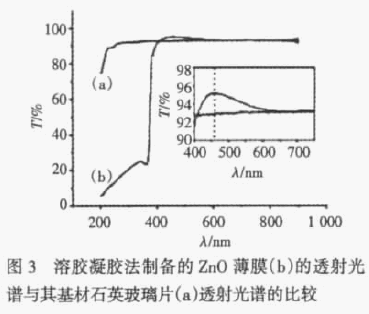
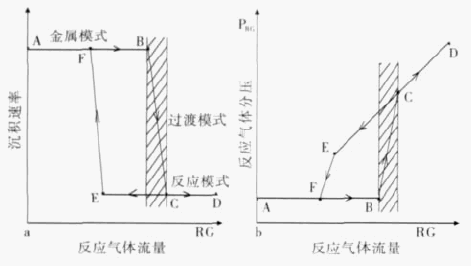
AlNį┌”┴-Al2O3(0001)▒Ē├µ╬³ĖĮ▀^│╠Ą─└Ēšō蹊┐
ĪĪĪĪ▓╔ė├╗∙ė┌├▄Č╚Ę║║»└ĒšōĄ─ŲĮ├µ▓©│¼▄ø┌Iä▌Ę©,ī”α-Al2O3 (0001) ▒Ē├µ╬³ĖĮAlN ▀Mąą┴╦äė┴”īW─ŻöMėŗ╦Ń,蹊┐┴╦AlNĘųūėį┌a-Al2O3 (0001) ▒Ē├µ╬³ĖĮ│╔µI▀^│╠Īó╬³ĖĮ─▄┴┐┼c│╔µIĘĮ╬╗ĪŻėŗ╦Ń▒Ē├„╬³ĖĮ▀^│╠Įø(j©®ng)Üv┴╦╬’└Ē╬³ĖĮĪó╗»īW╬³ĖĮ┼cĘĆ(w©¦n)Č©æB(t©żi)ą╬│╔Ą─▀^│╠,Ųõ╗»īWĮY║Ž─▄▀_ĄĮ4.844eVĪŻ╬³ĖĮ║¾AlN ╗»īWµI(0.189 ±0.010nm) ┼cūŅĮ³ÓÅĄ─▒Ē├µAl - O µIėą30°Ą─Ų½▐D(zhu©Żn)ĮŪČ╚,Al į┌▒Ē├µ▌^ĘĆ(w©¦n)Č©Ą─╗»īW╬³ĖĮ╬╗ų├š²║├Ų½ļx▒Ē├µO ┴∙ĮŪī”ĘQ╝s30°,╩╣Ą├AlN ┼c╦{īÜ╩»ų«ķgĄ─Š¦Ė±╩¦┼õČ╚ĮĄĄ═ĪŻ
ĪĪĪĪα-Al2O3 ,ėųĘQäéė±,ÅVĘ║Ąžė├ū„ųŲéõAlNĪóZnOĄ╚ļŖūė▒Ī─ż░ļī¦¾w▓─┴ŽĄ─╗∙Ų¼ĪŻ╦{īÜ╩»╗∙Ų¼╔Ž╔·ķLAlN ▒Ī─żį┌╬óļŖūėĪóļŖūėį¬╝■ĪóĖ▀ŅlīÆĦ═©ą┼ęį╝░╣”┬╩░ļī¦¾wŲ„╝■Ą╚ŅIė“Š▀ėąæ¬ė├Ū░Š░ĪŻŲõųą▓╔ė├Ęųūė╩°═Ōčė(MBE) Īó╝ż╣Ō├}ø_│┴Ęe(PLD) Īó╔õŅl┤┼┐ž×R╔õ│┴Ęe(MRS) Īó╬ó▓©Ą╚ļxūė▌oų·×R╔õĪó╚▄─z-─²─z(solgel) ĪóĮī┘ėąÖC╗»īWÜŌŽÓ│┴Ęe(MOCVD) Īó╝ż╣ŌĘųūė╩°═Ōčė(LMBE) Īó╝ż╣Ō╗»īWÜŌŽÓ│┴Ęe(LCVD) Ą╚šµ┐š╝╝ąg╔·ķLĄ─AlN ▒Ī─ż┘|(zh©¼)┴┐▌^Ė▀ĪŻ
ĪĪĪĪį┌▀@ĘĮ├µ,ĻPė┌AlN ▒Ī─żį┌α-Al2O3 (0001) ╗∙Ąū╔ŽĄ─ųŲéõĘĮĘ©┼c╔·ķL╠žąįėą▌^ČÓĄ─īŹ“×蹊┐ł¾ī¦,Č°ī”AlN ▒Ī─żį┌α-Al2O3 (0001) ▒Ē├µĄ─ūŅ│§╔·ķLÖC└Ē╔ą╚▒Ę”└Ēšōėŗ╦Ń蹊┐ĪŻ
ĪĪĪĪį┌šµ┐šųą,▓╔ė├═Ōčė╔·ķLĘ©ųŲéõ▒Ī─żĄ─╔·ķL▀^│╠ųą,╩ūŽ╚╗∙Ų¼Ą─▒Ē├µĮYśŗĪó╚▒Ž▌Ą╚ÅŖ┴꥞ė░Ēæ▒Ī─ż╔·ķLĄ──Ż╩ĮĪóą╬├▓ęį╝░Įń├µ╠žąį,Å─Č°ī”▒Ī─ż▓─┴ŽĄ─╣”─▄ŲĄĮ?j©®ng)QČ©ąįū„ė├ĪŻį┘ėą,│┴Ęe┴Żūėį┌╗∙Ų¼▒Ē├µ╬³ĖĮĪóöU╔óĪóĮY║Ž,ī”│╔║╦║═╔·ķL│§Ų┌ļAČ╬Ą─ąį┘|(zh©¼)ėąĘŪ│Żųžę¬Ą─ė░Ēæ,▓óų▒Įėė░Ēæų°īóꬹ╬│╔Ą─š¹éĆ▒Ī─żĄ─┘|(zh©¼)┴┐ĪŻė╔ė┌▒Ē├µīŹ“×Ęų╬÷╝╝ągī”▒ĒīėįŁūėĮYśŗĪó▒Ē├µµI║Ž┼c▒Ē├µļŖ║╔Īó▒Ē├µ╬³ĖĮęį╝░▒Ē├µä▌─▄Ą╚ĘĮ├µĄ─蹊┐╚į╚╗╚▒Ę”│õĘųĄ─īŹ“×蹊┐,ė╚Ųõ╩Ūī”ļsĄ─č§╗»╬’▒Ē├µĪŻę“┤╦▀\ė├┐╔┐┐Ą─└Ēšōėŗ╦Ń蹊┐Š¦¾w▒Ē├µĪó╝░Ųõ▒Ē├µĄ─╗»īW┼c╬’└Ē╬³ĖĮ,ęč│╔×ķę╗ĘNųžę¬Ą─Š¦¾w▒Ē├µčąŠ┐ĘĮĘ©ĪŻ
ĪĪĪĪ×ķ┴╦£pąĪ╩¦┼õČ╚Č°╠ßĖ▀AlN ▒Ī─żĄ─┘|(zh©¼)┴┐ęčĮø(j©®ng)ū÷┴╦┤¾┴┐Ą─蹊┐ ĪŻÅ─└Ēšō╔ŽüĒųv,AlN ┼c╦{īÜ╩»į┌c ▌S╔ŽĄ─Š¦Ė±╩¦┼õČ╚ĘŪ│Ż┤¾(61.11 %) ,Ą½īŹ“׳¾ī¦AlN ╚į╚╗┐╔ęįį┌╦{īÜ╩»╗∙Ąū╔Ž╔·ķL,▀@╩Ūė╔ė┌═©▀^╬’└ĒĘĮĘ©╩╣Ą├AlN Ž╚│╔║╦,╚╗║¾į┘╬³ĖĮį┌╗∙Ąū╔Ž├µ╗“š▀╩ŪŽ╚ūīGaN Ą╚ū÷ę╗éĆŠÅø_īė,╚╗║¾į┘╬³ĖĮĪŻ═¼ĢrČŁśõśsį┌FBAR ė├AlN ▒Ī─żĄ─╔õŅlĘ┤æ¬×R╔õųŲéõ蹊┐ę╗╬─ųąę▓╠ß│÷┴╦AlN ▒Ī─ż┐╔ęįŠ∙ä“Ą─čžc ▌SČčČŌ╔·ķLĪŻ╚╗Č°,ĄĮ─┐Ū░×ķų╣,į┌īŹ“×╔Ž▀ƤoĘ©Å─įŁūė│▀Č╚╔Ž½@Ą├┴Żūė╬³ĖĮ╔·ķL▀^│╠Ą─╬óė^äėæB(t©żi)ą┼Žó,╚ń╗∙Ąū▒Ē├µ╬’└ĒĪó╗»īW╬³ĖĮ╔·ķLĄ─▀^│╠,ęį╝░┴ŻūėūŅ│§į┌▒Ē├µĄ─╬³ĖĮ╬╗ų├ęį╝░▀\äė▄ē█EĪŻę“┤╦ī”▀@ą®ĘĮ├µ,ėą▒žę¬▀Mę╗▓Į蹊┐ĪŻ<
ĪĪĪĪ▒Š╬─ų„ę¬─┐Ą─╩Ū蹊┐AlN å╬Ęųūėį┌α-Al2O3(0001) ▒Ē├µĄ─╬³ĖĮ,ā╚(n©©i)╚▌░▓┼┼╚ńŽ┬:Ą┌1 ╣Ø(ji©”)║åę¬ĮķĮB┴╦╬’└Ē─Żą═┼cėŗ╦ŃĘĮĘ©,Ą┌2 ╣Ø(ji©”)ųž³cšō╩÷▓óėæšō┴╦AlN Ą─╬³ĖĮ▀^│╠┼c▒Ē├µĮYśŗ,ūŅ║¾Ą┌3 ╣Ø(ji©”)▀Mąą┴╦┐éĮYĪŻ
1Īó╬’└Ē─Żą═┼cėŗ╦ŃĘĮĘ©
ĪĪĪĪ╬ęéāų¬Ą└,AlN į┌α-Al2O3 (0001) ▒Ē├µöU╔óĪó╬³ĖĮ╬╗ų├╝░µI║Žū„ė├ĻPŽĄĄĮAlN ┴Żūė┤žąĪŹu▀Mę╗▓ĮĄ─ą╬│╔,ė░ĒæAlN ▒Ī─żĄ─╔·ķLĪŻę“┤╦蹊┐Ęų╬÷AlN į┌╗∙Ąū▒Ē├µĄ─╬³ĖĮ▀^│╠┼c│╔µI╠žąį,ī”蹊┐AlN ▒Ī─ż╔·ķL│§Ų┌Ą─ÖC└ĒėąĘŪ│Żųžę¬Ą─ęŌ┴xĪŻα-Al2O3 (0001) ▒Ē├µĄ─įŁūė┼c▒Ē├µļŖūėæB(t©żi)ī”═ŌüĒ┴ŻūėĄ─╬³ĖĮų▒ĮėŽÓĻP,×ķ┤╦, ╬ęéā╩ūŽ╚ėą▒žę¬Ą├ĄĮę╗éĆęč│┌įźĄ─α-Al2O3 (0001) ▒Ē├µ,Ęų╬÷Ųõ▒Ē├µįŁūė┼cļŖūėĮYśŗ,▀Mę╗▓ĮśŗįņAlN Ą─╬³ĖĮ─Żą═ĪŻ
ĪĪĪĪī”AlN Ą─╬³ĖĮ─Żą═,╗∙Ų¼▓╔ė├╬─½Ił¾ī¦ęčā×(y©Łu)╗»Ą─│¼Š¦░¹6 éĆįŁūėīėĄ─slab ▒Ē├µ(2 ×1) ─Żą═,▒Ē├µĮKų╣įŁūė×ķå╬īėAl Ą─▒Ē├µĮYśŗĪŻ×ķ┴╦ė^▓ņAlN ĘųūėųąĄ─N ║═Al ═¼╗∙Ų¼▒Ē├µĄ─Al ĪóO ĮY║ŽŽ╚║¾▀^│╠,╬ęéāīóā×(y©Łu)╗»Ą├ĄĮĄ─ÜŌæB(t©żi)AlN Ęųūė(µIŠÓ0.185nm) ,╦«ŲĮų├╚ļ╗∙Ų¼╔Ž┐š,ŠÓļx╗∙Ų¼▒Ē├µ013nm,│õĘų┐╝æ]┴╦šµ┐šģ^(q©▒)Ė▀Č╚▌^ąĪī”ėŗ╦ŃĄ─ė░Ēæ, ę“┤╦šµ┐šīėįOų├×ķ210nm,╚ńłD1(a) ĪŻŲõųą┐╝æ]┴╦▒Ē├µ╬³ĖĮ╬╗ų├Īó╬³ĖĮĘųūėĮYśŗ║═┐šųąŽ┬┬õĄ─ĘĮ╬╗ĪŻ×ķ┴╦▒Ē╩÷ĘĮ▒Ń,AlN ĘųūėųąĄ─N ═¼▒Ē├µĄ─Al ĮY║Ž▒Ē╩Š×ķ(Al)N - Al ,ŲõAl ═¼╗∙Ų¼▒Ē├µĄ─O ĮY║Ž,▒Ē╩Š×ķAl - O(╗∙Ų¼) ĪŻ
ĪĪĪĪα-Al2O3 (0001) ╗∙Ų¼▒Ē├µįŁūėĮYśŗ×ķč§6 ųžī”ĘQĮYśŗ,×ķ┴╦┐╝▓ņ▒Ē├µų„ę¬╬³ĖĮ╬╗ų├,įOČ©┴╦8 éĆ▓╗═¼Ą─ų„ę¬╬³ĖĮ╬╗ų├ ,▀@ą®╬╗ų├īŹļH╔Ž┐╔ęį┐┤ū„╩ŪAlN ▒Ī─żĄ─ūŅ│§╔·ķL³c,ęŖłD1 (b) ĪŻį┌(2 ×1) Ą─╬³ĖĮ─Żą═łD1 ųą,╬ęéāāH┐╝æ]┴╦AlN Ęųūė╦«ŲĮÅ─┐šųą┬õŽ┬Ą─ā╔ĘNŪķørĪŻę╗╩Ū─Żą═A:Al ĪóN Ęųäe╬╗ė┌╬³ĖĮ╬╗S7 ║═S6 ╔Ž┐š,╚ńłD1(c) ╦∙╩Š,AlN ĘųūėųąĄ─Al┼c▒Ē├µā╔éĆAl ŠÓļxŽÓĄ╚,Č°N ŠÓļx▒Ē├µAl ó┌▌^Į³ĪŻ┴Ēę╗ĘNŪķørŽÓĘ┤,╚ńłD1 (d) ─Żą═B ╦∙╩Š,N ┼c▒Ē├µā╔éĆAl ŠÓļxŽÓĄ╚,Č°AlN ĘųūėųąAl ŠÓļx▒Ē├µAl ó┌▌^Į³ĪŻ╬³ĖĮ─Żą═A ┼c─Żą═B ė├ė┌ė^▓ņAlN ĘųūėųąĄ─Al ║═N ŠÓļx▒Ē├µA1 ▀hĮ³ī”▒Ē├µ╬³ĖĮĄ─ė░ĒæĪŻ
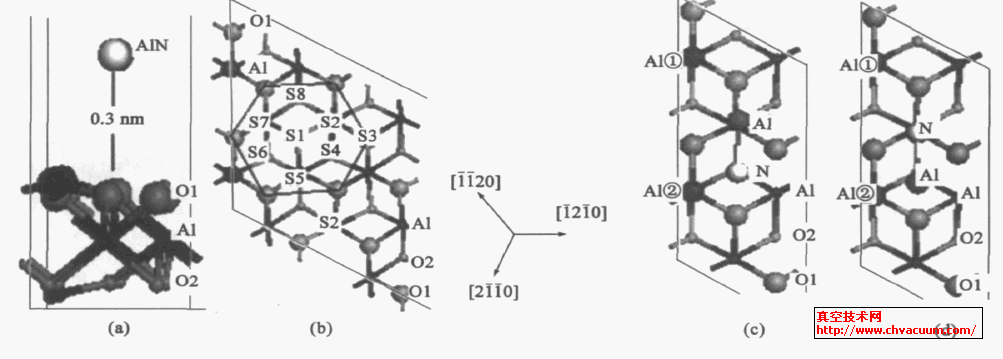
ĪĪĪĪłD1 ĪĪAlN/ a-Al2O3 (0001) ▒Ē├µ╬³ĖĮ╬╗ų├┼cAlN ╬³ĖĮ─Żą═(a) ▒ĪŲ¼╬³ĖĮ─Żą═, (b) Ēöīė▓╗═¼╬³ĖĮ╬╗ų├, (c) ╬³ĖĮ─Żą═A , (d) ╬³ĖĮ─Żą═BĪŻĪĪłDųą╗ę╔½ąĪŪ“┤·▒Ē╗∙Ų¼O įŁūė,ś╦×ķO2 ;║┌╔½ąĪŪ“┤·▒Ē╗∙Ų¼Al įŁūė;▌^┤¾Ą─║┌╔½Ū“┤·▒Ē╗∙Ų¼▒Ē├µĄ─ā╔éĆAl įŁūė,Ęųäeś╦ėø×ķAl ó┘║═Al ó┌;▌^┤¾Ą─╗ę╔½Ū“┤·▒Ē╗∙Ų¼O įŁūė,ś╦ėø×ķO1 ;░ū╔½ąĪŪ“┤·▒ĒN įŁūė,▌^┤¾Ą─╔Ņ╗ę╔½ąĪŪ“┤·▒ĒAlN ųąĄ─Al įŁūėĪŻ
ĪĪĪĪ╬ęéā▓╔ė├╗∙ė┌├▄Č╚Ę║║»└Ēšō( density functionaltheory ,DFT) ┐é─▄┴┐┌Iä▌Ę©,ė╔CASTEP(cambridge se2rial total energy package) ▄ø╝■░³īŹ¼F(xi©żn)ėŗ╦ŃĪŻ▀\ė├D.Vanderbilt ╠ß│÷Ą─│¼▄ø┌I(ultrasoftpseudopotentials ,USP) üĒ├Ķ╩÷ļxūėīŹ┼cārļŖūėų«ķgĄ─ŽÓ╗źū„ė├(O 2s22 p4 ,Al 3 s23 p1 ,N 2 s22 p5) ĪŻī”ļŖūėĮ╗ōQŽÓĻPĒŚĄ─ėŗ╦Ń▀xō±┴╦Perdew Ą╚╠ß│÷Ą─ÅV┴x╠▌Č╚Į³╦Ų(general gradient approximation ,GGA) ą▐š²ĘĮĘ©(PW91) ą╬╩ĮĪŻ═©▀^Ū░├µ┐éĮYĘų╬÷α-Al2O3 (0001) ▒Ē├µĄ─ėŗ╦Ń,╬ęéāų¬Ą└▀\ė├├▄Č╚Ę║║»└Ēšō,ŽÓī”ė┌ĮY║Ž─▄Ą─ėŗ╦ŃüĒšf,į┌ėŗ╦ѵIķL┼cÄū║╬śŗą╬Ģr,ī”ėŗ╦ŃŚl╝■ŽÓ«ö▓╗├¶Ėą,ļŖ║╔├▄Č╚Ęų▓╝ļSėŗ╦ŃŚl╝■Ą─ūā╗»ŽÓ«öąĪ,ŽÓī”Š∙ĘĮ▓Ņį┌10 - 4┴┐╝ē,╠žäeī”ė┌öĄ(sh©┤)ųĄĘeĘų³cöĄ(sh©┤)▓╗├¶Ėą ĪŻ×ķ┴╦▀m«ö£pąĪäė┴”īWĄ─▀\╦Ń┴┐,į┌▓╔ė├│¼▄ø┌Iä▌ėŗ╦ŃĢr,┐╔ęį▀m«ö£pąĪĮžöÓ─▄EcutĄ─╚ĪųĄ,╬ęéā?n©©i)ĪĮžöÓ─▄┴┐Ecut×ķ340eVĪŻį┌ī”2×2 ▒Ē├µĮYśŗĄ─ā×(y©Łu)╗»ųą,▓╔ė├┴╦BFGS ╦ŃĘ©(Broyden Fletcher Goldfarb and Shanno ,BFGS) ,▓╝└’£Yģ^(q©▒)k2point ╚ĪĄ─╩Ū3 ×3 ×2 ,╝┤18 éĆK-point ĪŻī”2 ×1 ▒Ē├µAlN Ą─╬³ĖĮäė┴”īW─Żą═,╬ęéā▀\ė├┴╦CASTEP ▄ø╝■░³ųąĄ─äė┴”īW│╠ą“▓╝└’£Yģ^(q©▒)k-point ╚ĪĄ─╩Ū5 ×2 ×1 ,╝┤10 éĆk-point ,ĘeĘųĢrķg▓ĮķLįO×ķ1.0 fs ,─ŻöM┐éĢrķg×ķ1.5ps ,¾wŽĄ£žČ╚║ŃČ©×ķ500 ĪµĪŻ═©▀^ī”α-Al2O3(0001) ▒Ē├µĮYśŗā×(y©Łu)╗»┼c│┌įźėŗ╦Ń,ęį╝░ī”AlN ĘųūėµIķLĪóµI─▄įć╦Ń,ĮY╣¹Č╝ĘŪ│Ż║├Ąž╬Ū║Žė┌╬─½IųĄ ,═¼Ģr┼cGauss98 ▄ø╝■░³╦∙ėŗ╦ŃĄ─AlN µIķLųĄ(0.170nm) ę▓ĘŪ│Żę╗ų┬,▒Ē├„╬ęéāėŗ╦ŃĮY╣¹╩Ū┐╔┐┐Ą─ĪŻ